BOSTON, APRIL 7, 2026 —
Key Takeaways
- Columbia University researchers are running a landmark ALS gene therapy trial in which patients receive spinal infusions every few months that target and silence the mutated gene responsible for their form of ALS — a disease that typically kills within 2 to 5 years of diagnosis
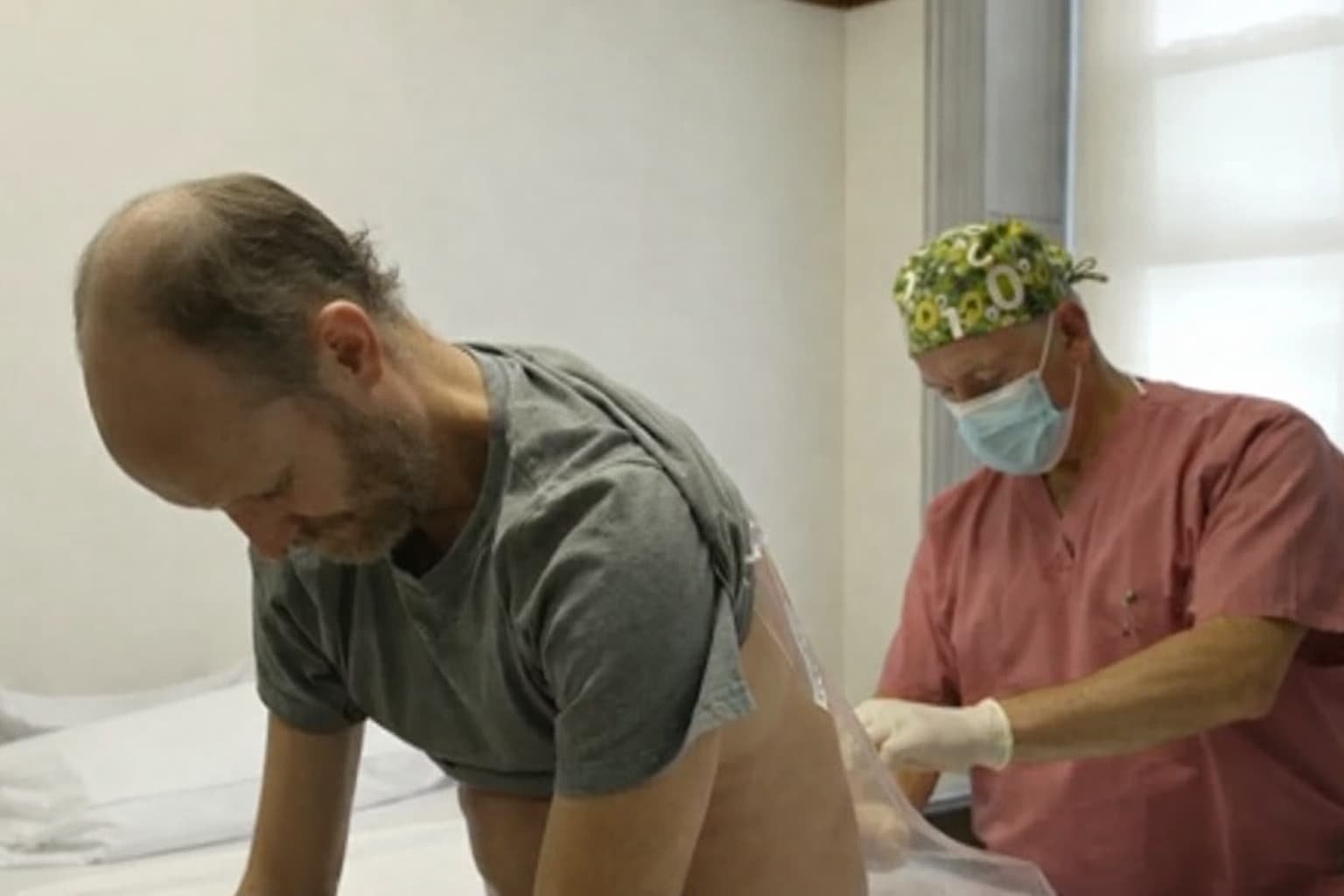
- At least one patient — Jeff Vierstra, featured this week by CBS News — has been receiving the treatment for three years with no measurable disease progression, representing an outcome almost unheard of in ALS history
- The therapy is currently limited to patients with the SOD1 mutation, which accounts for roughly 2% of ALS cases — but researchers say the approach could be adapted for other genetic forms of the disease affecting a much larger population
ALS — amyotrophic lateral sclerosis, also called Lou Gehrig’s disease — is one of the cruelest diagnoses in medicine. The disease destroys the motor neurons that control movement, speech, swallowing, and eventually breathing. It has no cure. The median survival after diagnosis is two to five years. The disease does not care how old you are, how fit you are, or who you are — it killed baseball legend Lou Gehrig at 37, physicist Stephen Hawking at 76, and hundreds of thousands of ordinary Americans in between.
What happened to Jeff Vierstra in the last three years is, by the standards of ALS medicine, almost incomprehensible.
What the Treatment Is
Vierstra has a form of ALS caused by a mutation in the SOD1 gene — one of several known genetic mutations that cause inherited ALS. SOD1 mutations account for roughly 2% of all ALS cases and approximately 20% of familial ALS cases (ALS that runs in families). The mutation causes the SOD1 protein to misfold, becoming toxic to motor neurons.
The therapy Vierstra has been receiving is a type of antisense oligonucleotide (ASO) treatment — a class of drugs that use short synthetic DNA-like molecules to bind to and silence a specific gene’s messenger RNA, preventing the production of the harmful protein. The drug is delivered via spinal infusion — an injection into the cerebrospinal fluid that surrounds the brain and spinal cord — every few months.
The goal is not to reverse damage already done. It is to halt the production of the toxic protein before it can destroy more motor neurons — essentially putting the disease on pause.
According to researchers at Columbia University running the Silence ALS initiative, the approach is working in Vierstra’s case in a way that has not been seen before in ALS treatment: after three years of infusions, his disease has shown no measurable progression.
Why This Matters — The History of ALS Treatment
ALS research has had very few genuine breakthroughs in its history. The FDA has approved several treatments — riluzole in 1995, edaravone in 2017, tofersen in 2023 for SOD1-ALS — but most have provided only modest slowing of decline rather than the kind of sustained stability Vierstra appears to be experiencing.
Tofersen — the most recent SOD1-targeting drug, marketed as Qalsody by Biogen — was the first drug specifically designed for SOD1-ALS and uses the same antisense oligonucleotide mechanism. Clinical trial data showed it reduced neurofilament light chain levels — a biomarker of nerve damage — significantly compared to placebo, and slowed functional decline in some patients. The Columbia Silence ALS approach appears to be taking a similar but more individualized path, designing therapies specific to each patient’s mutation.
ALS — The Scale of the Disease in America
| Statistic | Figure |
|---|---|
| Americans diagnosed with ALS annually | ~5,000 |
| Americans living with ALS at any time | ~30,000 |
| Median survival after diagnosis | 2–5 years |
| 5-year survival rate | ~10% |
| SOD1 mutation cases | ~2% of all ALS, ~20% of familial ALS |
| FDA-approved ALS treatments | 4 (riluzole, edaravone, tofersen, relyvrio — though relyvrio was withdrawn in 2024) |
| Cost of tofersen (Qalsody) annually | ~$197,000 |
What “Silence ALS” Is and How to Participate
The Silence ALS initiative at Columbia University Irving Medical Center is developing individualized gene-based therapies for patients with rare genetic forms of ALS. The program focuses on creating patient-specific ASO treatments for mutations that are too rare for large pharmaceutical companies to prioritize in standard drug development pipelines.
Patients or family members interested in the Silence ALS initiative can write to silenceals@cumc.columbia.edu for information about potential participation.
The program represents a model of precision medicine for rare diseases — where instead of developing one drug for many patients, researchers design individual therapies for each patient’s specific genetic variant. The approach is expensive and complex, but for patients with no other options, it represents the frontier of what is now possible.
The Cautious View
One patient doing well for three years is not a clinical trial. Anecdotal results — even extraordinary ones — require replication in larger, controlled studies before they can be generalized. The ALS field has seen promising early results collapse in later trials before.
What is undeniably true is that the tools now available to researchers — gene silencing, antisense oligonucleotides, CRISPR-based editing, individualized medicine platforms — represent a genuine step change from what was possible a decade ago. For the roughly 5,000 Americans diagnosed with ALS every year, the pace of that progress is a matter of life and death.



